Study Guide
Chondroblastoma
Key Points:
- Chrondroblastoma is a benign tumor of bone with slow and self-limited growth, 1% of all bone tumors
- Commonly seen in the epiphyses and apophyses of long bones in adolescents and young adults; Males > Females (2:1)
- Chrondroblastomas are derived from cells of tendon sheaths that abut the epiphyseal plate and undergo intraosseous proliferation to form a tumor
- Positive for S100 stain.
- Key histologic finding is “chicken-wire calcification”.
- The recurrence rate is 10-15% after standard treatment with intralesional curettage and packing with allograft bone chips.
- Differential Diagnosis: giant cell tumor of bone, chondromyxoid fibroma, and clear cell chondrosarcoma
Description:
Chondroblastomas are benign bone tumors that make up about 1% of all bone tumors. They were initially classified by Jaffe and Lichtenstein in 1940s, who renamed them from their original designation as “chondromatous variant of giant cell tumor”, given by Codman in 1931 (Codman, 1931; Jaffe, 1942). They are typically characterized by slow and self-limited growth and are located in the epiphysis or apophysis of long bones.Epidemiology:
Chondroblastomas typically occur in adolescents and young adults. According to the largest studies, the average age is 22 years old (Ramappa, 2000). They occur more frequently in men than women at a 2:1 ratio and are found most commonly in the epiphyses and apophyses of long bones, primarily the proximal tibia and humerus (Atalar, 2007). Cases of chondroblastoma have also been reported in the pelvis, femur, calcaneus, talus, scapula, patella, radius, metatarsals, skull, and fibula.Clinical Findings:
The most common presentation of chondroblastoma is localized pain. The pain is often mild and progressive, and can sometimes be confused with minor trauma occurring in the past. It can be accompanied by limitations of movement, a palpable mass, or an effusion. These secondary characteristics are dependent upon the location of the tumor. Those with intra-articular involvement are most likely to cause limitations in motion and effusions. These symptoms are sometimes mistaken for inflammatory joint disease in younger patients, especially when an effusion is involved (Atalar, 2007).Imaging Studies:
Initial workup begins with history and physical examination, followed by imaging of the affected joint or bone. Radiographs will show an osteolytic lesion, most commonly in the epiphyseal plate. The lesion is well-circumscribed. Radiographs will show a well-circumscribed, osteolytic lesion, most commonly in the epiphysis. There may be physeal involvement, and 20% will show a hemorrhagic or cystic lesion (Cozzolino, 2015). Other potential findings may include expansion of bone and a sclerotic margin. If questions remain regarding the diagnosis or extent of the lesion, CT or MRI of the area may be performed, which should show characteristic calcifications that may be difficult to see on plain radiographs. (Figure 1) Bone scan may also be performed in cases where the biologic activity of the lesion is unclear. Chest x-ray should be obtained as benign pulmonary metastases have been documented in the past.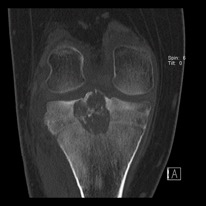
Figure 1: CT of chondroblastoma located in the proximal tibia
Photo courtesy of Rajiv Rajani, MD Department of Othopedics UTHSCSA
In cases where clinical suspicion for malignancy is low, biopsy may be performed at the time of surgical treatment in order to confirm diagnosis. Otherwise, a core needle or incisional biopsy with histologic confirmation should be done prior to definitive treatment.
Differential diagnosis includes giant cell tumor of bone (GCT), chondromyxoid fibroma (CMF), and clear cell chondrosarcoma. GCT has a similar bone distribution to chondroblastoma, but occurs later in life, usually in skeletally mature patients. Histologically GCT will show absence of calcifications and presence of clustered, elongated cells. If histologic appearance does not confirm a diagnosis, an S100 stain can be performed which will be positive in chondroblastoma. Clear cell chondrosarcoma differs in that it will show higher degrees of calcification and will stain positive for type II collagen. CMF typically has a different distribution, with a higher preference for the metadiaphysis of long bones (Romeo, 2009). Histologically, the chondroblastoma lesion will show well-defined chondroblasts with benign-appearing nuclei, multinucleated giant cells, chondroid islands, and patterns of small calcific bodies surrounded by chondroblasts, also known as “chicken-wire calcification.” (Figure 2) These findings should be present in all chondroblastomas. Other histologic findings that can appear in chondroblastoma include pseudonucleoli, foam cells, and sporadic calcifications. These findings should not be considered necessary for diagnosis (Cozzolino, 2015).
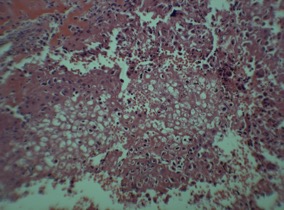
Figure 2: Histologic image of chondroblastoma showing chicken-wire calcification
Photo courtesy of Rajiv Rajani, MD Department of Orthopedics UTHSCSA
Treatment:
Surgery is the primary and definitive treatment for chondroblastoma. Intralesional curettage and packing with allograft bone chips is the most common treatment. Alternatively, polymethylmethacrylate may be used for larger tumors. Some studies show a potential decrease in rates of recurrence with its use instead of allograft, although no statistical significance has been noted. Radiation and chemotherapy have no current role in treatment of chondroblastoma (Atalar, 2007). Adjuvants can be utilized to decrease the approximately 10-15% recurrence rate. These include phenol, argon, peroxide, alcohol, and high-speed burr.Recent efforts to find medical therapy for cases in which surgery is not an option have shown some success. A study in 2013 by Yang, et al discussed the potential of mTOR and HIF as targets of inhibition which have been shown to slow tumor growth (Yang, 2013).
Complications:
Because there is a risk for local recurrence, patients should be monitored for several years after treatment. Monitoring should include a full history, physical exam and repeat radiographs. Patients with open physes at the time of treatment should be monitored for premature physeal arrest. Most cases proceed without complication following surgical therapy. Osteoarthritis may develop as a result of both treatment and joint collapse. Epiphyseal tumors in an open growth plate may results in limb-length discrepancy and/or angular deformity.There is a 10% risk of recurrence, on average 34 months following surgery. Recurrence is thought to be related to both the aggressiveness of the tumor and inadequate surgical technique. No statistical significance has been associated with any of the supposed risks of recurrence (young age, location of the tumor, aggressiveness of tumor). Physeal-sparing surgery was shown not to increase the risk of recurrence. Recurrence can be treated with repeat curettage or en bloc resection and reconstruction, depending on the size and location (Atalar, 2007; Yang, 2013; Lin, 2005).
In addition to recurrence, there have been cases of metastatic disease reported. In case reports, outcomes vary significantly, from death to absence of symptoms. Another subset of chondroblastomas may undergo malignant transformation. This is usually discovered years after initial treatment and confers a poor prognosis (Ostrowski, 1999).
Related Videos:

References:
- Atalar H, Basarir K, Yildiz Y, Erekul S, Saglik Y. Management of chondroblastoma: retrospective rview of 28 Patients. J Orthop Sci. 2007; 12(4): 334-340.
- Brien, EW., Mirra JM, Ippolito V. Chondroblastoma arising from a nonepiphyseal site. Skeletal Radiol. 1995;24 (3).
- Codman EA. The classic: epiphyseal chondromatous giant cell tumors of the upper end of the humerus. Surg Gynecol Obstet.1931; 52:543.
- Cozzolino I, Zeppa P, Zabatta A, Merolla F, Vetrani A, Sadile F. "Benign chondroblastoma on fine-needle aspiration smears: a seven-case experience and review of the literature. Diagn. Cytopathol. 2015;43(9) :734-738.
- Jaffe HL, Lichtenstein L. Benign chondroblastoma of bone: a reinterpretation of the so-called calcifying or chondromatous giant cell tumor. Am J Pathol. 1942:18:969-991.
- Lin PP, Thenappan A, Deavers MT, Lewis VO, Yasko AW. Treatment and prognosis of chondroblastoma. Clinical Orthop Relat Res. 2005; 438:103-09.
- Ostrowski ML, Truong LD, Johnson ME, Spjut HJ, Hicks MJ., Malignant chondroblastoma presenting as a recurrent pelvic tumor with DNA aneuploidy and P53 mutation as supportive evidence of malignancy. Skeletal Radiology 1999:28(11): 644-650.
- Ramappa AJ, Lee FY, Tang P, Carlson JR, Gebhardt MC, Mankin HJ. Chondroblastoma of bone. J Bone Joint Surg 2000; 82-A( 8): 1140-1145.
- Romeo, S, Hogendoorn PC, DeTos AP. Benign cartilaginous tumors of bone. Advances in Anatomic Pathology 2009;16(5) 307-315.
- Yang, X, Yang ZJ, Liu FX, et al. Inhibition of MTOR and HIF pathways diminishes chondro-osteogenesis and cell proliferation in chondroblastoma. Tumor Biol. 2013;.5: 3111-3119.
Top Contributors:
Dr. KleinRajiv Rajani MD